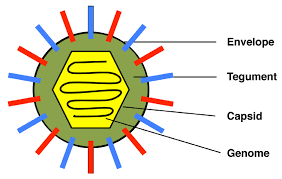
Summary
Many enveloped viruses are released from infected cells by maturing and budding at the plasma membrane. During this process, components of the viral core are incorporated into membrane vesicles that contain viral transmembrane proteins, called “spike” proteins. For many years, these spike proteins, which are required for infectivity, were believed to be incorporated into virions through direct interaction between their cytoplasmic domains and components of the viral core.
More recent evidence shows that while such direct interactions drive budding in alphaviruses, this may not be the case for negative-stranded RNA viruses and retroviruses. These viruses can generate particles in the absence of spike proteins, using only viral core components to drive the process. In some cases, the spike proteins, without the viral core, can be released as virus-like particles. Therefore, optimal sprouting and release may depend on a concerted ‘push and pull action of the core and spike, where oligomerization of both components plays a crucial role.
As viruses are obligate intracellular pathogens, they cannot replicate without the machinery and metabolism of a host cell. Although the replicative life cycle of viruses differs greatly between virus species and categories, there are six basic stages that are essential for viral replication.
1. Adhesion: Viral proteins on the capsid or phospholipid envelope interact with specific receptors on the host cell surface. This specificity determines the host range (tropism) of a virus.
2. Penetration: The process of binding to a specific receptor can induce conformational changes in the viral capsid proteins, or the lipid envelope, resulting in fusion of the viral and cellular membranes. Some DNA viruses can also enter the host cell through receptor-mediated endocytosis.
3. Stripping: The viral capsid is removed and degraded by viral enzymes or host enzymes that release the viral genomic nucleic acid.
4. Replication: After the viral genome has been unwrapped, transcription or translation of the viral genome begins. It is this stage of viral replication that differs greatly between DNA and RNA viruses and viruses with opposite nucleic acid polarity. This process culminates in the de novo synthesis of viral proteins and genome.
5. Assembly: After de novo synthesis of the viral genome and proteins, which can be modified post-transcriptionally, the viral proteins are packaged with the newly replicated viral genome into new virions that are ready to be released from the host cell. This process can also be called maturation.
6. Virion release: There are two methods of viral release: lysis or budding. Lysis results in the death of an infected host cell, these types of viruses are called cytolytic. One example is variola major, also known as smallpox. Enveloped viruses, such as the influenza A virus, are usually released from the host cell by budding. It is this process that results in the acquisition of the phospholipid viral envelope. These types of viruses do not usually kill the infected cell and are called cytopathic viruses.
After virion release, some viral proteins remain within the host cell membrane, serving as potential targets for circulating antibodies. Residual viral proteins that remain within the host cell cytoplasm can be processed and presented at the cell surface on MHC class I molecules, where they are recognized by T cells.
Materials And Methods
- Viruses and cells.
Vero and HeLa cells were maintained in Dulbecco’s Modified Eagle’s Medium (Invitrogen) supplemented with 5% fetal bovine serum, 5% bovine serum, penicillin and streptomycin. Virus stocks were prepared by infecting confluent monolayers of Vero cells with the KOS strain of HSV type 1 or the Becker strain of pseudorabies virus (PRV) at a multiplicity of infection of 0.01 for 1 h.

After infection, cells were incubated in Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum, 25 mM HEPES buffer, glutamine (0.3 μg/ml), penicillin, and streptomycin until a cytopathic effect was visualized. complete (~3 days). Cells were then scraped into the medium, frozen (-80 °C) and thawed (37 °C) three times, and then sonicated (4 °C) at moderate power for three pulses of 1 min. Cellular debris was removed by centrifugation and aliquots were frozen at -80°C until use.
- Mutant viruses.
The GC-null mutant and revertant (GC-R) used in these studies were propagated in Vero cells as described previously (kindly provided by Harvey Friedman). The GB null mutants (KO82 and F-BAC GB-) and complementary cells (VB38) used for their growth were generously provided by David Johnson. To generate GB-deficient virions, the progeny of GB-depleted viruses were obtained after infection of non-complementary Vero cells. The ΔUL16 mutant used in the supporting studies (see Supplementary Material) was a gift from Joel Baines and has been described previously.
- MEN treatment of herpesviruses.
Extracellular virions harvested between 18 and 24 h post-infection were treated with 10 mM NEM for 30 min at 37 °C, either before or after unwrapping with NP-40 (final concentration, 0.5 %. ; I3021; Sigma). Previous work revealed that this concentration and treatment time efficiently modify free cysteines, and in our system also maintain the UL16-capsid interaction in virions.
After NEM treatment, virions and capsids were pelleted at 83,500 × g for 1 hour and samples were separated on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels and electroblotted onto nitrocellulose membranes. The enhanced chemiluminescence method of immunoblot analysis was performed according to the manufacturer’s instructions (Amersham). Rabbit anti-HSV UL16 and anti-VP5 sera were used at dilutions of 1:6,000 and 1:7,500, respectively. PRV UL16-specific antibodies were kindly provided by Thomas Mettenleiter and used at a dilution of 1:15,000.
- Virus-cell binding assay.
Confluent monolayers of cells in 100-mm dishes were incubated with 1 mL of virus stock (∼5 × 108 PFU) at 4°C with rocking for 45 min to allow virus attachment. NEM was added to the virus before or after incubation with cells for 30 min at 4°C. Unbound virus and residual NEM were removed by washing the cells with 5 ml of phosphate-buffered saline (PBS).
Viral membranes that remained attached to the cells were solubilized with 4 mL of NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl [pH 8.0]) for 15 min. at 37°C. Lysates were removed from the dish by pipetting, and insoluble material and cell debris were removed by centrifugation for 10 min at 3,829 × g. The supernatant capsids were pelleted through a 700 μl 30% sucrose cushion (wt/vol in TNE [20 mM Tris-HCl {pH 7.6}, 100 mM NaCl, 1 mM EDTA]) in a rotor. SW55ti at 83,500 × g for 1 h.
Pellets were dissolved in SDS sample buffer (2% SDS, 62.5 mM Tris-HCl, 10% glycerol, 0.025% bromophenol blue, 50 mM dithiothreitol [DTT], and 5% β-mercaptoethanol [BME]), boiled and separated by SDS. – polyacrylamide gel electrophoresis (PAGE) in 10% gels, and analyzed by immunoblotting using specific antibodies for VP5 and UL16. The amount of UL16 was determined by densitometry and normalized to VP5 levels.